Dose Selection in Proof-of-Concept Trials: When to use a single “no regret” dose versus multi-dose evaluation
In this piece, Dr Simon Hutchings, Senior Clinical Pharmacologist at Weatherden shares his thoughts and experiences regarding the advantages and disadvantages of evaluating a single dose compared to multiple dose levels when designing PoC clinical studies".
1. Introduction
The decision to proceed in a proof-of-concept (PoC) trial with a single “no-regret” dose (NRD) versus a multi-dose evaluation has profound implications for resource use, interpretability, and risk.
A single NRD approach can accelerate timelines and reduce cost, but risks misinterpreting negative results, e.g. if a supratherapeutic dose results in a safety signal or a non-monotonic (‘bell-shaped’) exposure-response. A multi-dose design provides richer exposure–response data and supports regulatory justification and dose optimization, but at greater operational complexity and cost.
Emerging adaptive and model-based approaches offer hybrid pathways that seek to combine the strengths of both strategies. This paper reviews the advantages and disadvantages of each approach, including regulatory, statistical, clinical, and operational considerations, and provides recommendations for choosing the appropriate design in various development contexts.
2. Role of Dose Selection in Early Clinical Development
Dose selection is a critical lever in drug development. Poor dose choice in early phase clinical trials is a major contributor to late-stage failure.
Several reviews and analyses of drug development attrition attribute a considerable proportion of Phase 3 trial failures to suboptimal dose selection(1–4). For example, the failure of gantenerumab to slow clinical decline in patients with Alzheimer’s disease was attributed to insufficient therapeutic exposure, and that evaluating higher doses earlier in development may have prevented this outcome.
Regulatory expectations reflect this risk: the FDA’s Exposure-Response Relationships guidance emphasizes that dose- or concentration-response relationships are central to determining safe and effective dosing and should be considered throughout development(5). Similarly, the ICH E4 guidance (Dose-Response Information to Support Drug Registration) underlines the importance of exploring dose-response relationships early in development in order to guide later dose selection(6).
In practice, drug developers must balance competing priorities: speed, cost, interpretability, regulatory acceptability, and risk mitigation. That balance underlies the trade-off between NRD designs and multi-dose evaluations.
3. Regulatory and Strategic Contexts
Regulatory Expectations and Guidance
As noted, the FDA’s Exposure–Response Relationships guidance encourages integrated planning for exposure–response analysis across development, and recommends that sponsors prospectively plan dose-response study designs (e.g. randomized fixed-dose studies)(5). The ICH E4 guidance also underscores the value of dose–response information in selecting dose and dosage regimens(6).
In oncology specifically, Project Optimus reflects the FDA’s evolving stance: sponsors are encouraged to conduct randomized dose comparisons and to move away from defaulting to MTD-based designs(7). This represents a regulatory push toward more deliberate dose optimization earlier in development.
Several recent oncology-focused publications argue for reforming dose-finding practices, recommending that dose optimization become part of early-phase development and that designs incorporate clinical activity signals, not only toxicity(8–10). For example, the recent article Totality of the Evidence: Optimizing Dosage Selection Strategies in Oncology outlines the need for paradigm shifts in dose-finding practices(10).
Dose optimization is increasingly seen as essential not only to efficacy but also to tolerability, especially for targeted agents that may have narrower differentiation at lower doses(9–11). A recent review by Glasmacher et al. emphasizes that clinical trials should characterize the dose–response curve and identify a dose range early(11).
The FDA’s Optimizing the Dosage of Human Prescription Drugs and Biological Products guidance also underscores that dose-finding trials should evaluate a range of dosages and leverage exposure–response data to guide selection(12). If a dosage is chosen without adequate justification, the FDA may require additional bridging or dose-ranging trials before approval.
Strategic Considerations for Sponsors
Regulatory alignment: early engagement with regulatory authorities can clarify whether a NRD design is acceptable or whether they will require additional dose-response data. It should be noted that regulatory acceptability of a NRD design for an individual trial is different to the regulatory acceptability of an NRD approach supporting dose selection in later phase trials/application for marketing authorisation.
Risk tolerance: a sponsor with high risk tolerance might accept the possibility of a false negative to expedite development, while a more conservative sponsor may prefer more robust multi-dose designs. Risk tolerance may also vary between different regulatory authorities.
Therapeutic area norms: in chronic disease or therapeutic areas where dose–response is poorly predicted, multi-dose designs are often expected.
Commercial differentiators: dose flexibility, titration, or labelling advantages (including paediatric considerations) may favour multi-dose data.
Portfolio sequencing: in pipeline planning, one program may absorb more risk to move quickly (NRD) while others take a more measured approach.
4. The Single “No-regret dose” Proof-of-Concept Design: Concept and Rationale
A NRD design is an attempt to optimize the trade-off between efficacy, exposure and tolerability.
A NRD design refers to choosing a single dose level for Phase 1b/Phase 2 Proof-of-Concept (PoC) trials such that the sponsor is unlikely to “regret” the choice, i.e. the dose is anticipated to result in maximal pharmacodynamic effect, and has a credible chance of efficacy without excessive safety risk. It is an attempt to optimize the trade-off between efficacy, exposure and tolerability based on prior (preclinical / Phase 1) data/modelling, avoiding the cost and complexity of testing multiple arms.
This concept is more common in therapeutic areas where effect sizes are expected to be large, where patient recruitment is constrained (e.g. oncology, rare genetic and metabolic disorders), or where there is high uncertainty regarding the mechanism and therefore a higher perceived risk of failure.
Advantages of the Single NRD Strategy
1. Operational Simplicity and Speed
With a single dose level, the logistics of site activation, randomization, dose preparation, monitoring, and data management are simpler. There is no need to manage multiple dose arms (e.g. different packaging, blinding across dose levels). This can shorten start-up time and reduce operational burden.
2. Cost Containment
By eliminating or reducing the need for multiple dose arms, sponsors can reduce sample size, monitoring complexity, and overhead costs. For programs with tight budgets or early go/no-go inflection points, the cost savings may be compelling.
3. Clear Decision Rule and Simpler Statistical Interpretation
A trial with a single dose level simplifies the decision criterion: either the dose succeeds (meets prespecified efficacy criteria) or it does not. There is no need to infer which dose is best or to adjust for multiple comparisons among dose arms.
This simplicity aids decisional clarity, particularly when the hypothesis is “does this mechanism work at a feasible exposure” rather than “what is the optimal dose.”
Risks and Limitations of a Single NRD Strategy
Insufficient Characterization of the Exposure–Response Relationship
Perhaps the greatest risk is that with only one dose the trial provides no internal evidence of how efficacy and safety vary with exposure. If the selected dose fails, there is no contingency of another dose to rescue a positive signal.
Without multiple dose levels, one cannot determine whether the failure was due to an inadequate dose (i.e. “too low”) or to lack of pharmacologic activity. Where failure is due to toxicity, how far above a safe/tolerated dose the NRD selected was cannot be determined. In dose–response terms, one cannot delineate the slope, plateau, or shape of the curve from a single point.
Increased Chance of Type II (False Negative) Outcome
If the NRD is suboptimal, even by a modest margin, the trial might falsely conclude there is no signal. This is particularly risky in contexts where inter-subject variability or PK/PD uncertainty is high.
Perception of Higher Risk and Reduced Confidence from Stakeholders
Investors, partners, or external evaluators may view a single-dose level PoC as higher risk because the data do not support dose optimization. The limited evidence may weaken commercial or partnering negotiations.
Difficulty Handling Heterogeneity or Subpopulations
If response or exposure differs across subpopulations (e.g. renal impairment, body size, genetic subgroups), a single dose level may under- or over-expose certain subgroups, thereby increasing the risk of failed efficacy or unacceptable toxicity, respectively.
5. Multi-Dose Phase 2 Designs (Dose-Ranging / Dose-Finding): Advantages and Challenges
Definition and Rationale
In a multi-dose design, the sponsor tests multiple dose levels (often greater than 2) within the PoC trial to map the exposure–response relationship. This allows estimation of efficacy and safety as functions of dose and exposure, which supports selection of the optimal dose for subsequent development (Phase 2b/Phase 3) and regulatory submissions.
While dose-ranging or dose-finding based on efficacy/pharmacodynamic effect is a long-established paradigm in non-oncology drug development (e.g. in cardiovascular, metabolic, CNS drug therapeutic areas), in recent years it has received increasing emphasis in the oncology space due to evolving regulatory expectations (see “Project Optimus” below).
Advantages of Multi-Dose Evaluation
Rich Exposure–Response Characterization
By evaluating multiple dose levels, one can determine the slope, plateau, or curvature of the dose–response relationship, identify the minimum effective dose (MED), and locate the region of maximal benefit with acceptable risk. This supports robust dose selection for Phase 3.
Dose–response modelling also enables simulation, interpolation, and extrapolation to untested higher or
intermediate doses, thereby refining dose choice.
Mitigation of False Negative Risk
If one dose fails (e.g. too low or unexpectedly poorly tolerated), other doses may still deliver a positive signal. This redundancy reduces the risk of discarding a viable development path due to mis-specified dose.
Regulatory Credibility and Labelling Support
Regulatory agencies (FDA, EMA) generally favour designs that generate dose–response evidence. The FDA’s exposure–response guidance explicitly states that exposure–response or dose–response data can help support dose and regimen selection and labelling(5). The ICH E4 guidance similarly positions dose–response studies as critical to registration dossiers(6). In oncology, FDA’s Project Optimus initiative signals a shift toward expecting randomized comparisons among dose levels rather than defaulting to the maximum tolerated dose (MTD) philosophy(7).
Multi-dose data also help support dose modifications in subpopulations and inform benefit-risk trade-offs.
Reduced Need for Later Bridging Studies
Because exposure–response has already been explored, sponsors are less likely to need subsequent dose-ranging or bridging studies before launching pivotal trials. This front-loads development risk but may shorten the overall timeline.
Clarity in Dose Comparison and Decision Support
When multiple doses are evaluated concurrently, direct statistical comparisons enable decision-making (e.g. is dose A significantly better than dose B, or statistically significant dose/exposure relationship). Such comparative data strengthen internal and external confidence in the chosen dose.
Challenges, Risks, and Trade-Offs
Increased Complexity and Logistics
Multiple dose arms require more complex trial infrastructure: multiple formulations or strengths, randomization stratification, blinding across arms and dosing logistics. The complexity of execution and data handling scales with the number of arms.
Greater Sample Size and Cost
To maintain statistical power across multiple arms, more patients are required per dose. The overall trial cost may increase by 50 to 100% or more, depending on the number of arms and the effect sizes assumed. Importantly, the correct dose range must still be robustly selected and justified.
Distribution of Power Across Arms
If the total sample size is fixed (e.g. due to budget constraints, or prevalence of a rare disease), splitting patients across multiple dose arms reduces per-arm sample size, thereby diluting power to detect differences; especially in contexts with high interpatient variability or modest effect sizes.
Operational and Statistical Risk of Multiple Comparisons
Testing multiple doses introduces multiplicity concerns (e.g. inflation of type I error). Appropriate statistical adjustments or hierarchical modelling must be applied, further complicating analysis. In some designs, sequential adaptation or Bayesian borrowing must be carefully controlled to maintain integrity.
Potential for Slower Decision Making
With more arms and more data to assess, timelines to reach a conclusive decision may be extended compared to a single-dose level readout, particularly if interim decisions are planned.
6. Comparative Summary
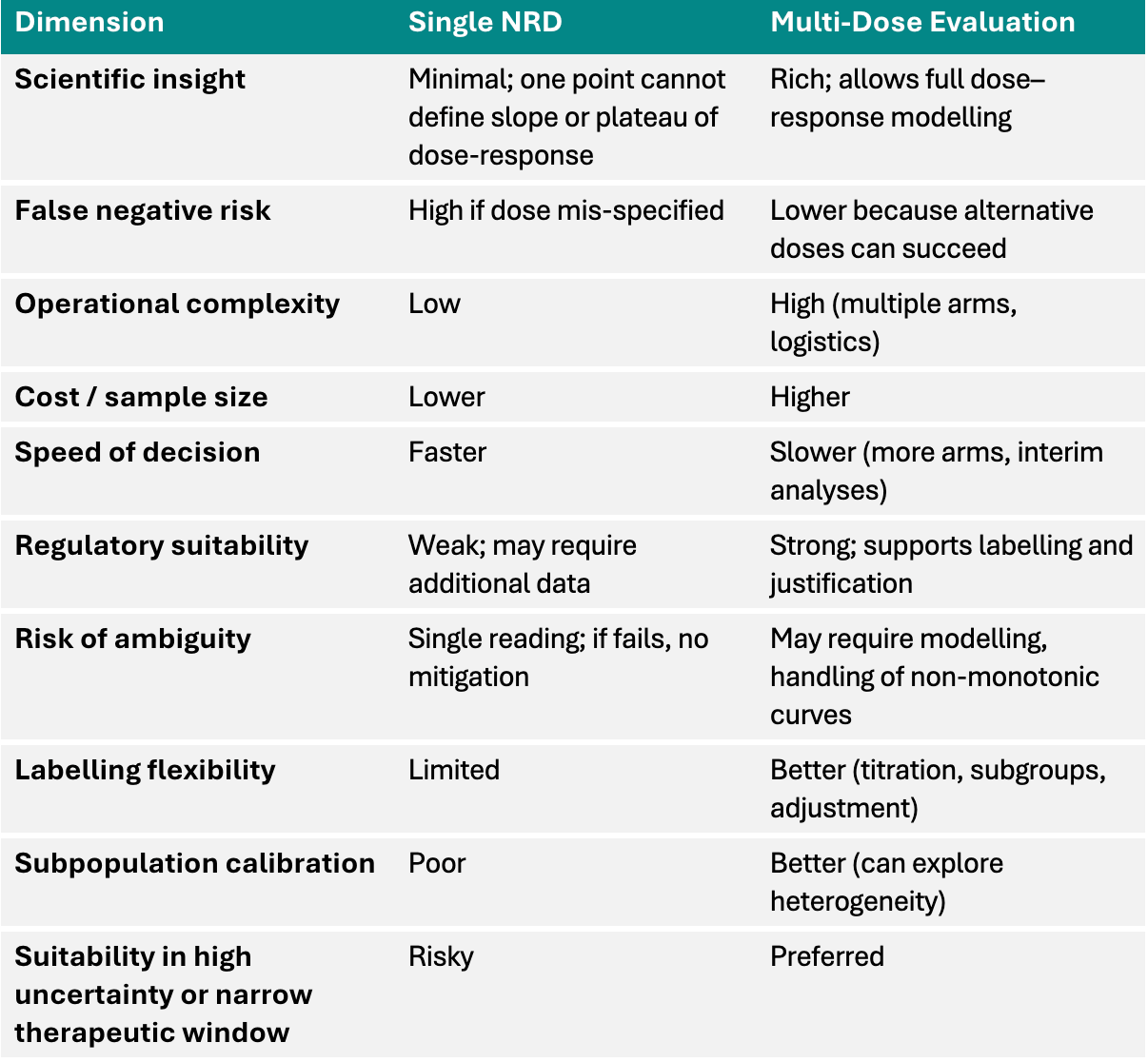
Make it stand out
This aligns with guidance from the FDA and industry practice recommending evaluation of a range of doses during development and justifying dose selections with exposure-response evidence(5,6,12). The oncology literature also strongly supports early dose optimization rather than reliance on MTD alone(9–11).
7. Hybrid and Adaptive Designs: Blending Efficiency and Rigor
Emerging approaches aim to bridge the gap between the simplicity of single NRD designs and the richness of multi-dose designs. Some prominent strategies include:
1. Adaptive Dose-Finding / Model-Based Designs
Adaptive designs dynamically allocate patients across dose arms in response to interim results, concentrating enrolment toward more promising doses. Bayesian adaptive randomization and model-based designs (such as Continual Reassessment Method, CRM, or Bayesian hierarchical models) can improve efficiency and reduce the number of patients exposed to suboptimal doses.
For example, the Multi-Arm Two-Stage (MATS) design has been proposed for oncology PoC and optimization: it begins with two selected doses, enrols patients to both, and then in a second stage randomizes further patients to the more promising doses while dropping less promising ones(13). This design allows simultaneous proof-of-concept and dose optimization. Similarly, others have proposed MERIT, DROID, or seamless randomized multiple-dose approaches in oncology to align with Project Optimus goals(14–16).
These adaptive strategies reduce the sample size and duration compared to fully factorial static designs, while maintaining exposure–response learning.
2. Seamless Phase 2/3 Dose-Optimization Designs
Inferentially seamless trial designs integrate dose-finding and confirmatory evaluation in one protocol. In stage 1 (Phase 2), multiple doses are randomized; in stage 2 (Phase 3), the selected dose(s) continue in a confirmatory setting without restarting a separate trial. Simulation studies suggest that seamless dose-optimization designs can reduce sample size requirements (e.g. 16–27 % savings) while controlling type I error rates(15). However, they demand careful planning of interim decision rules, control of error rates, and operational rigor.
3. Model-Informed Drug Development (MIDD) and Simulation
Quantitative pharmacology and mechanistic modelling can support dose selection even in a NRD setting, by leveraging preclinical and Phase 1 PK/PD data to estimate the exposure–response shape and simulate response under alternative doses. Regulatory agencies are increasingly open to model-based justification of dosing regimens(5,12). When modelling confidence is high, a NRD may be defensible.
Moreover, clinical trial simulators can forecast operating characteristics (power, probability of success) under various designs, aiding rational planning.
4. Borrowing Across Indications / Populations via Hierarchical Models
In programs spanning multiple indications or subpopulations, Bayesian hierarchical or borrowing models can share exposure–response information across arms, reducing the need for large per-arm sizes. The MATS design mentioned earlier uses hierarchical modelling to borrow strength between doses and indications(13).
8. Case Illustrations and Literature Examples
Oncology: Movement toward Dose Optimization (Project Optimus)
FDA’s Project Optimus aims to shift oncology development away from the “more is better / MTD” paradigm toward dose optimization strategies that balance efficacy and safety(7). Consistent with this aim, several recent oncology reviews advocate integrating activity data, biomarker endpoints and dose-finding within early phases(8–10). As an example, a 2024 commentary by Hoog et al. (Dose selection of novel anticancer drugs: exposing the gap) argues for more rational dose selection using biomarker, PK/PD, and multi-arm evaluation(17).
Hansen et al. previously proposed that expansion cohorts testing multiple schedules/doses beyond MTD can be useful to yield early activity insights and support estimation of dose-response relationships in an efficient manner(18).
Dose Optimization in Oncology Trials
The recent review by Glasmacher et al. (Dose optimization in cancer drug development) underscores the need to characterize dose–response early and to randomize among dose options rather than defaulting to MTD(11). For example Liao et al. (14) highlight the different relationships between optimal dose and the MTD when comparing traditional cytotoxic therapy, molecular targeted agents and immunotherapy (Figure 1). Other works propose novel designs: e.g. DROID (dose-ranging approach for oncology adaptive trials) and MERIT as frameworks for dose optimization beyond classic escalation(14,16). The use of MATS as an integrated PoC plus dose-optimization design offers proof-of-concept and dose comparison in a unified trial(13).
Seamless Dose-Optimization
The seamless Phase 2/3 dose-optimization approach demonstrates, via simulation, sample size efficiencies (16–27 % reduction) compared to conventional separate trials(15). This illustrates the operational benefit of combining dose-finding and confirmatory phases.
General Drug Development and Regulatory Approaches
FDA’s guidance Project Optimus initiative illustrates how dose-finding trials should evaluate multiple doses and justify choices using exposure–response evidence(12). The FDA’s Exposure–Response Relationships guidance further clarifies expectations around prospective planning of exposure–response studies, including dose–response and concentration–response analyses(5). Interestingly, the response by the global pharmacometrics leader, Certara, to the FDA / CDER internal oncology dose optimization docket further argues for use of population PK, E–R modelling, and randomization among doses or subpopulations(19).
The ICH E4 guideline (and companion documents) remain foundational for dose–response information in registration(6). For example, the ICH E4 text states that “knowledge of the relationships among dose, drug concentration … and clinical response” is crucial to safe and effective dosing in patients.
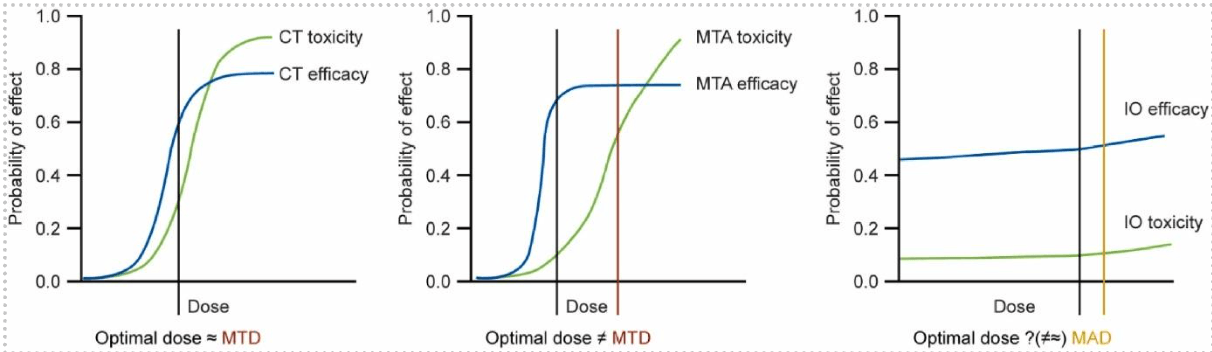
Fig. 1. Relationship between the optimal dose and the MTD/MAD among different drug treatments. Note: CT = cytotoxic therapy; IO = immunotherapy; MAD = maximum administered dose; MTA = molecularly targeted agent; MTD = maximum tolerated dose. Reproduced from Liao et al. (14)
9. Recommendations and Best Practices
When a Single NRD Makes Sense (with caveats)
A single-dose PoC study may be defensible when:
The mechanism of action is well understood and the pharmacology-translational bridge is strong.
Preclinical/Phase 1 data (e.g. PK, target engagement, biomarker response) provide confidence in exposure–response relationship.
The therapeutic window is wide (i.e. safety margin is large), reducing risk of toxicity surprises.
The disease area is one of high unmet-need, or includes specialised populations (e.g. rare disease, oncology), where speed is crucial.
The sponsor is willing to accept higher risk (including likelihood of follow-up dose-range finding studies).
Regulatory engagement confirms that a single-dose PoC would be acceptable for the indication.
In such cases, a model-based justification may strengthen the case for a single dose level. However, sponsors must be prepared to defend the lack of dose-ranging data in regulatory review.
When Multi-Dose Designs (Or Hybrid Approaches) Are Preferred
Multi-dose or adaptive approaches are more appropriate when:
Exposure–response uncertainty is substantial (e.g. high intersubject variability, unknown pharmacodynamics).
The therapeutic index is narrow, making precise dose calibration essential.
Label differentiation, titration, or subpopulation dosing is desired.
The program can absorb higher upfront cost for reduced downstream risk.
Regulatory expectations or competitor standards favour robust dose justification.
Modelling confidence is low and mitigation/flexibility is needed.
Utilising adaptive or Bayesian dose-finding designs (e.g. MATS, DROID, MERIT) helps balance resource use and learning. Seamless designs may further reduce sample sizes while preserving dose optimization integrity (subject to rigorous design)(13,15,16).
Design and Statistical Best Practices
Allocate sufficient sample size per dose arm to detect meaningful differences; avoid spreading patients too thinly across many doses.
Predefine adaptive decision rules (dose dropping, expansion) with appropriate control of type I and II error.
Use population PK/PD or exposure–response modelling concurrently to guide dose adjustment and interpretation.
Leverage borrowing or hierarchical modelling across subpopulations or indication arms when possible.
Simulate operating characteristics (power, type I error, probability of correct dose identification) for candidate designs prior to selection.
Plan interim analyses carefully and control for multiplicity or Bayesian prior influence.
Engage regulators early to align on dose-response expectations, adaptation rules, and coverage of labelling claims.
Monitor emerging safety/efficacy continuously and be ready to adapt if assumptions fail.
10. Future Directions
Continuous learning systems and adaptive dose escalation/de-escalation are likely to become standard, reducing the dichotomy between single-dose and multi-dose.
AI/machine-learning-augmented simulation platforms may better forecast design performance under uncertainty.
Regulatory evolution (e.g. further expansion of Project Optimus principles beyond oncology) may raise expectations for dose optimization in all therapeutic areas.
Real-time biomarker-driven adaptive dosing may allow patient-by-patient dose titration within trials, enabling dynamic exposure–response learning.
Post-marketing dose-optimization studies may become more accepted as part of lifecycle management, especially when initial dosing was conservative.
11. Conclusion
Choosing between a single NRD and a multi-dose Phase 2 design is not a purely technical decision but a strategic one, balancing speed, cost, interpretability, and risk.
The single-dose design offers simplicity and speed but carries a higher risk of misspecification and limited regulatory defensibility. The multi-dose approach offers richer insight and confidence but at greater complexity and expense.
Emerging hybrid and adaptive designs (e.g. MATS, DROID, seamless Phase 2/3) and quantitative pharmacology methods help bridge the gap. The optimal strategy depends on translational confidence, therapeutic window, regulatory expectations, and organizational risk appetite. Wherever possible, sponsors should simulate candidate designs, engage regulators early, and embed exposure–response modelling in the development plan.
Curious about how our Pharmacology team can fit into your strategy? Get in touch to learn how our experts can give your drug the best chance of success in clinical trials.
12. References
Ogasawara K, Breder CD, Lin DH, Alexander GC. Exposure– and Dose–response Analyses in Dose Selection and Labeling of FDA-approved Biologics. Clin Ther. 2018;40(1):95-102.e2.
2. Yap C, Rekowski J, Ursino M, Solovyeva O, Patel D, Dimairo M, et al. Enhancing quality and impact of early phase dose-finding clinical trial protocols: SPIRIT Dose-finding Extension (SPIRIT-DEFINE) guidance. BMJ. 2023; 383:e076386.
3. Pretorius S, Grignolo A. Phase III Trial Failures: Costly, But Preventable. Appl Clin Trials. 2016;25(8). Available from: https://www.appliedclinicaltrialsonline.com/view/phase-iii-trial-failures-costly-preventable
4. Buyse M, Saad E. Reasons Behind Phase 3 Clinical Trials Failure IDDI. 2024. Available from: https://iddi.com/resources/why-do-so-many-phase-3-trials-fail
5. US FDA. Exposure-Response Relationships - Study Design, Data Analysis, and Regulatory Applications. 2003. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/exposure-response-relationships-study-design-data-analysis-and-regulatory-applications
6. ICH. E4 Dose response information to support drug registration - Scientific guideline. 1994. Available from: https://www.ema.europa.eu/en/ich-e4-dose-response-information-support-drug-registration-scientific-guideline
7. US FDA. Project Optimus. 2024. Available from: https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus
8. Jaki T, Burdon A, Chen X, Mozgunov P, Zheng H, Baird R. Early phase clinical trials in oncology: Realising the potential of seamless designs. Eur J Cancer. 2023;189:112916.
9. Almuradova E, Izzo D, Gandini S, Gaeta A, Giordano E, Valenza C, et al. From Dose-Finding to Dose-Optimization in Early-Phase oncology clinical trials. Cancer Treat Rev. 2025;136:102906.
10. Levit LA, Shah M, Ratain MJ, Garrett-Mayer E, Rahman A, Theoret M, et al. Totality of the Evidence: Optimizing Dosage Selection Strategies in Oncology. Journal of Clinical Oncology. 2025;43(25):2827–33.
11. Glasmacher A, Garralda E, Gwaltney C, Rupalla K, Li C, Weber H. Dose optimization in cancer drug development: Review and outcome of a multi-stakeholder workshop. Eur J Cancer. 2025;226:115593.
12. US FDA. Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases Guidance for Industry. 2024. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/optimizing-dosage-human-prescription-drugs-and-biological-products-treatment-oncologic-diseases
13. Jiang Z, Mi G, Lin J, Lorenzato C, Ji Y. A Multi-Arm Two-Stage (MATS) Design for Proof-of-Concept and Dose Optimization in Early-Phase Oncology Trials. 2023; Available from: https://arxiv.org/pdf/2304.06164
14. Liao JJZ, Asatiani E, Liu Q, Hou K. Three steps toward dose optimization for oncology dose finding. Contemp Clin Trials Commun. 2024;40:101329.
15. Jiang L, Yuan Y. Seamless Phase 2-3 Design: A Useful Strategy to Reduce the Sample Size for Dose Optimization. 2022; Available from: https://arxiv.org/pdf/2211.02046
16. Guo B, Yuan Y. DROID: Dose-ranging Approach to Optimizing Dose in Oncology Drug Development. Biometrics. 2022;79(4):2907–19.
17. Hoog CJPO, Mehra N, Maliepaard M, Bol K, Gelderblom H, Sonke GS, et al. Dose selection of novel anticancer drugs: exposing the gap between selected and required doses. Lancet Oncol. 2024;25(8):e340–51.
18. Hansen AR, Cook N, Amir E, Siu LL, Abdul Razak AR. Determinants of the recommended phase 2 dose of molecular targeted agents. Cancer. 2017;123(8):1409–15.
19. Bullock J, Chassereau F, Duval V, Koeck K, Manon A, Poggesi I, et al. Response to ‘Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases’. 2023. Available from: https://downloads.regulations.gov/FDA-2022-D-2827-0007/attachment_1.pdf